The Musculoskeletal System
The Musculoskeletal System
-
The skeleton is made of cartilage and bone.
-
Cartilage helps in growth and repair of bone and forms joints.
-
Bone provides support, movement, and acts as a calcium reservoir.
-
Human body contains 206 bones which can be long, flat, or tubular.
Normal Structure of Bone
Two Types of Bone:
-
Cortical (Compact) Bone – 80% of skeleton
-
Dense outer shell
-
Strong and rigid
-
Contains osteons with Haversian canals
-
-
Trabecular (Cancellous) Bone – 20% of skeleton
-
Spongy with bone trabeculae
-
Involved in mineral balance
-
Histology of Bone
Bone Cells and Matrix:
-
Osteoblasts
-
Bone-forming cells
-
Produce bone matrix (osteoid)
-
Marker: ↑Alkaline Phosphatase
-
Active in growth, fracture healing
-
-
Osteocytes
-
Mature osteoblasts trapped in matrix
-
Reside in lacunae
-
Help maintain bone
-
Woven vs. Lamellar Bone:
-
Woven bone = immature, random collagen (children, fractures)
-
Lamellar bone = mature, organized collagen (adults)
-
-
-
Osteoclasts
-
Multinucleated, bone-resorbing cells
-
Origin: Macrophages
-
Found in Howship’s lacunae
-
Marker: ↑Acid Phosphatase
-
-
Osteoid Matrix
-
90–95% collagen type I
-
Contains hydroxyproline, hydroxylysine
-
Can be woven or lamellar depending on synthesis rate
-
Bone Formation and Resorption
-
Bone is dynamic, constantly remodeled by:
-
Osteoblasts – Form bone (deposition)
-
Osteoclasts – Break bone (resorption)
-
-
Ossification types:
-
Membranous Ossification: Direct from collagen (e.g. skull bones)
-
Endochondral Ossification: Via cartilage stage (e.g. long bones)
-
Bone Matrix Formation:
-
Starts with uncalcified osteoid
-
Mineralized in 12–15 days
-
Mineralized matrix stains black (von Kossa (special histological staining method used to detect calcium deposits)), unmineralized is eosinophilic
Normal Structure of Cartilage
-
Avascular: No blood vessels, nerves, or lymphatics
-
Components:
-
Cartilage Matrix
-
80% water
-
20% collagen type II & proteoglycans (glycosaminoglycans: chondroitin sulfate & keratan sulfate)
-
Functions: Lubrication, shock absorption
-
-
Chondrocytes
-
Derived from mesenchymal cells
-
Mature cartilage cells
-
-
Types of Cartilage
-
Hyaline Cartilage
-
Most common
-
Found in joints, nose, trachea, growth plates (epiphyseal plate—is a hyaline cartilage plate located between the epiphysis and metaphysis of long bones in growing individuals)
-
Seen in fracture callus and cartilage tumors (Cartilage tumors are neoplastic proliferations of chondrocytes)
-
-
Fibrocartilage
-
Hyaline cartilage with extra collagen II
-
Found in intervertebral discs, ligaments (dense regular connective tissue structures that connect bone to bone)
-
-
Elastic Cartilage
-
Hyaline cartilage + elastin
-
Found in ear, epiglottis, larynx
-
Diseases of Skeletal System
-
Infections – e.g. osteomyelitis
-
Growth disorders – skeletal dysplasias
-
Metabolic diseases – e.g. rickets, osteoporosis
-
Tumors – benign and malignant bone/cartilage tumors
OSTEOMYELITIS
Definition:
Osteomyelitis is an infection of the bone and bone marrow. It may occur due to haematogenous spread, direct inoculation, or extension from adjacent infections.
I. TYPES OF OSTEOMYELITIS
A. Pyogenic Osteomyelitis (Bacterial)
-
Causative Organisms:
-
Most common: Staphylococcus aureus
-
Others: Streptococci, E. coli, Pseudomonas, Klebsiella, anaerobes
-
Mixed infections: Post-traumatic cases
-
-
Age Group Affected:
-
Common in children aged 5–15 years, particularly in developing countries
-
-
Routes of Infection:
-
Haematogenous – more common in children
-
Direct Extension – from adjacent infected tissues (common in jaws and skull)
-
Direct Inoculation – compound fractures, surgery, implants
-
-
Risk Factors:
-
Diabetes with gangrene
-
Immunosuppression
-
Debilitating conditions
-
Clinical Features
-
Painful, tender limb
-
Fever, malaise, leukocytosis
-
Bony destruction evident on radiographs
-
May remain undiagnosed → becomes chronic
-
Chronic cases may develop:
-
Draining sinuses
-
Secondary squamous cell carcinoma
-
Secondary amyloidosis (extracellular deposition of abnormal, misfolded proteins)
-
Morphologic Stages and Features
-
Acute Osteomyelitis
-
Infection starts in metaphysis
-
Pus in marrow cavity → increased pressure
-
Spread to endosteum, haversian and Volkmann canals
-
Periosteitis
-
-
Subperiosteal Abscess
-
Pus extends under periosteum
-
May penetrate cortex → sinus tracts to skin
-
-
Sequestrum Formation
-
Ischemic necrosis of cortical bone due to pressure and infection
-
Sequestrum = dead bone fragment
-
-
Involucrum Formation
-
New bone forms under periosteum
-
Involucrum = sheath of new bone encasing the sequestrum
-
Contains perforations for sinus tracts
-
-
Chronic Osteomyelitis
-
Dense sclerotic bone seen
-
Known as Garré’s osteomyelitis
-
-
Brodie’s Abscess
-
Localized, walled-off infection in bone
-
Surrounded by granulation and fibrous tissue
-
-
Vertebral Osteomyelitis
-
Begins as discitis
-
Spreads to vertebral bodies
-
Complications of Pyogenic Osteomyelitis
-
Septicaemia
-
Acute bacterial arthritis
-
Pathological fractures
-
Squamous cell carcinoma in sinus tracts
-
Secondary amyloidosis
-
Vertebral collapse, cord compression (in spinal involvement)
II. Tuberculous Osteomyelitis
-
Causative Agent: Mycobacterium tuberculosis
-
Route: Haematogenous spread from lungs or GIT
-
Common Sites:
-
Vertebrae (Pott’s disease)
-
Long bones of extremities
-
Joints and synovium
-
-
Histopathologic Features:
-
Caseating granulomas
-
Central necrosis surrounded by tuberculous granulation tissue
-
Necrotic bone fragments
-
Multiple sinus tracts
-
Special Forms
-
Pott’s Disease:
-
Tuberculous spondylitis
-
Vertebral body destruction
-
Collapse and compression fractures
-
May lead to paraplegia
-
-
Psoas Abscess:
-
Extension of infection from lumbar vertebra to psoas muscle sheath
-
Forms a lumbar cold abscess
-
May discharge via skin sinus
-
-
Complications:
-
Permanent vertebral damage
-
Paraplegia (partial or complete loss of motor and/or sensory function in both lower limbs)
-
Secondary amyloidosis
-
Summary Chart
| Feature | Pyogenic Osteomyelitis | Tuberculous Osteomyelitis |
|---|---|---|
| Cause | Bacteria (S. aureus) | M. tuberculosis |
| Route | Haematogenous, direct extension | Haematogenous from lungs/GIT |
| Site | Long bones (metaphysis) | Spine, long bones |
| Histology | Neutrophilic infiltration, necrosis | Caseating granulomas |
| Special Features | Sequestrum, Involucrum, Sinus tracts | Pott's disease, Psoas abscess |
| Complications | Septicaemia, fractures, carcinoma | Paraplegia, amyloidosis |
AVASCULAR NECROSIS (OSTEONECROSIS)
Definition:
Bone death due to ischemia, especially in end-arterial areas.
Etiology:
-
Fracture/dislocation
-
Sickle cell disease
-
Corticosteroids
-
Radiation
-
Alcoholism
-
Idiopathic
Pathogenesis:
-
Interruption of blood flow by trauma, compression, or thromboembolism
Morphology:
-
Infarcts mainly in medulla of long bones (diaphysis)
-
Subchondral infarcts under joint surfaces
-
Microscopy: Saponified marrow fat (breakdown of necrotic adipocytes in bone marrow, where fatty acids combine with calcium salts, forming chalky, white, soap-like deposits); cortex/cartilage often spared
Complications:
-
Pathologic fractures
-
Secondary malignant tumors: Osteosarcoma, fibrosarcoma
SKELETAL DYSPLASIAS (Disorders of Bone Growth and Development)
Local Defects:
-
Absence/hypoplasia of bones
-
Fusion (e.g., syndactyly)
-
Supernumerary bones
Systemic Disorders:
Involve the epiphyseal growth plate and include:
1. Achondroplasia
-
Autosomal dominant
mutation in the fibroblast growth factor receptor 3 (FGFR3) gene that results in its protein being overactive.
-
Defective endochondral ossification (long bones)
-
Normal skull size; short stature
-
Most common cause of dwarfism
2. Osteogenesis Imperfecta (Brittle Bone Disease)
-
Genetic disorder (dominant or recessive)
-
Defective Type I collagen synthesis
-
Affects bones and other collagen-rich tissues
-
Fragile bones, blue sclerae, hearing loss, abnormal teeth
-
Forms:
-
Congenita: At birth, severe
-
Tarda: Adolescence, milder
-
3. Osteopetrosis (Marble Bone Disease)
-
Autosomal recessive (malignant) or dominant (benign)
-
Defective osteoclast function → excess dense bone
-
Marrow space obliteration → anemia, neutropenia, hepatosplenomegaly
-
Neurologic symptoms: Deafness, blindness
-
Histology: Increased, bizarre, non-functional osteoclasts
Metabolic and Endocrine Bone Diseases
These are a group of disorders that affect the entire skeleton due to systemic metabolic or hormonal imbalances. Major examples include:
1. Osteoporosis
-
Definition: Quantitative reduction of otherwise normal bone mass, leading to increased fragility.
-
Common in: Elderly and postmenopausal women.
-
Clinical Features:
-
Often asymptomatic.
-
Backache, fractures (distal radius, femoral neck, vertebrae).
-
-
Radiology:
-
Changes visible only after >30% bone loss.
-
Normal serum calcium, phosphate, and ALP.
-
-
Types:
-
Primary:
-
Idiopathic: In young/juveniles (rare).
-
Involutional: Postmenopausal/aging (common).
-
-
Secondary:
-
immobilization: Mechanical loading is essential for bone maintenance. ↓ Osteoblastic activity,↑ Osteoclastic resorption. Examples: Long-term bed rest, paralysis, space travel
chronic anemia: especially hemolytic and thalassemic states) leads to Marrow expansion → thinning of cortical bone. ↑ Osteoclastic resorption due to increased marrow demands
endocrine disorders (hyperparathyroidism, hyperthyroidism): ↑ PTH → Stimulates osteoclasts. Excess thyroid hormone →Preferential increase in bone resorption
medications: corticosteroids (↓ Osteoblast formation and function, ↑ Osteoclast lifespan, ↓ Calcium absorption (gut), ↑ renal calcium excretion), anticonvulsants (↑ Vitamin D catabolism → ↓ calcium), heparin (Direct osteoclast activation (long-term use)).
-
-
-
Pathogenesis:
-
Imbalance between bone resorption and formation.
-
Risk Factors: Genetics, sex (female), physical inactivity, hormone deficiencies (estrogen, androgen), low vitamin D, local bone turnover factors.
-
-
Morphology:
-
Active osteoporosis: ↑ osteoclasts & osteoblasts, normal osteoid seam width.
-
Inactive osteoporosis: ↓ bone turnover, ↓ osteoclasts and osteoblasts, reduced osteoid.
-
2. Osteomalacia and Rickets
-
Cause: Vitamin D deficiency → impaired bone mineralization.
-
Osteomalacia: In adults.
-
Rickets: In children.
3. Scurvy
-
Cause: Vitamin C deficiency (cofactor for prolyl and lysyl hydroxylase)
-
Effect: Impaired collagen synthesis → subperiosteal hemorrhages.
| Pathological Process | Explanation |
|---|---|
| ↓ Hydroxylation of collagen | Leads to formation of defective, unstable collagen → weak connective tissue and capillaries |
| Fragile capillaries | Prone to rupture → bleeding into tissues, especially under the periosteum |
| ↓ Osteoid formation | Poor matrix formation → defective bone structure |
| ↓ Wound healing | Due to impaired collagen deposition in granulation tissue |
4. Osteitis Fibrosa Cystica (OFC)
🔄 Pathogenesis Overview
Osteitis Fibrosa Cystica is a skeletal manifestation of hyperparathyroidism, driven by excess parathyroid hormone (PTH), which increases osteoclastic bone resorption, leading to bone loss, fibrosis, cyst formation, and brown tumors.
Mechanism: Role of Excess PTH
| PTH Action | Effect on Bone |
|---|---|
| Stimulates osteoblasts | Indirectly ↑ RANKL expression |
| RANKL binds to RANK on osteoclast precursors | Promotes osteoclast differentiation and activity |
| ↑ Osteoclastic resorption | Leads to bone demineralization and matrix degradation |
| Bone microenvironment damage | Results in fibrosis, microfractures, and hemorrhage |
Types of Hyperparathyroidism
| Type | Cause | ||
|---|---|---|---|
| Primary | Parathyroid adenoma (most common), hyperplasia | ||
| Secondary | Chronic renal failure → hypocalcemia → ↑ PTH |
Morphological Stages
1. Early Stage
-
Subperiosteal bone resorption (classic feature)
-
Seen especially in phalanges
Thinning or disappearance of the lamina dura in the jaw
-
2. Intermediate Stage
-
Widespread osteoclastic activity
-
Marrow spaces replaced by fibrous tissue
-
Called “osteitis fibrosa” (fibrosis of bone)
3. Advanced Stage
-
Formation of cystic lesions containing:
-
Hemorrhage
-
Multinucleated giant cells
-
Fibrous tissue
-
-
These are called “brown tumors”
-
Brown color due to hemosiderin deposition
-
Not true neoplasms
-
Histologically resemble giant cell reparative granulomas
-
Common Sites Involved
| Long bones | |
| Jaw | |
| Vertebrae |
🧪 Biochemical Profile (Primary Hyperparathyroidism)
| Parameter | Level |
|---|---|
| Calcium | ↑ (hypercalcemia) (Increases calcium reabsorption in the distal tubules) |
| Phosphate | ↓ (hypophosphatemia) (Decreases phosphate reabsorption in the proximal tubules) |
| PTH | ↑ |
| Alkaline phosphatase | ↑ (due to high bone turnover) |
| Urinary calcium | ↑ (hypercalciuria) |
Clinical Features
| Symptom | Explanation |
|---|---|
| Bone pain | Due to microfractures and periosteal stress |
| Skeletal deformities | Bone loss and remodeling |
| Pathologic fractures | Weakened structure due to osteolysis |
| Brown tumor masses | Can be palpable if near the surface |
5. Pituitary Dysfunctions
-
Hyperpituitarism:
-
↑ GH → Gigantism (before epiphyseal closure) and Acromegaly (after closure).
-
-
Hypopituitarism:
-
↓ GH → Dwarfism.
-
6. Thyroid Dysfunctions
-
Hyperthyroidism: Causes osteoporosis (↑ bone turnover).
-
Hypothyroidism: In children causes cretinism (growth retardation) (delayed endochondral ossification)
7. Renal Osteodystrophy
-
Occurs in: Chronic renal failure (CRF), dialysis patients.
-
Clinical Signs: Usually subclinical; evident on X-ray/histology.
-
Pathogenesis:
-
Hyperphosphatemia (due to ↓ renal excretion) → hypocalcemia → ↑ PTH → secondary hyperparathyroidism.
-
↓ Vitamin D activation → ↓ calcium absorption.
-
Metabolic acidosis → bone resorption.
-
Aluminium toxicity (from dialysis) → inhibits mineralization.
-
β2-microglobulin accumulation → dialysis-related amyloidosis.
-
-
Skeletal Manifestations:

8. Skeletal Fluorosis
-
Cause: Excess fluoride (from soil/water).
-
Effect: Dense but brittle bones; joint stiffness and pain.
Summary Table: Key Differences
| Disease | Main Cause | Bone Mineral Content | Histology | Serum Calcium | Unique Features |
|---|---|---|---|---|---|
| Osteoporosis | Aging, inactivity, estrogen loss | ↓ | Thin trabeculae, normal osteoid | Normal | Fragility fractures |
| Osteomalacia/Rickets | Vit D deficiency | ↓↓ (due to poor mineralization) | ↑ Osteoid seams | Low/normal | Looser zones, soft bones |
| Osteitis Fibrosa Cystica | Hyperparathyroidism | Variable | Fibrosis, giant cells, brown tumors | ↑ | Bone resorption, cysts |
| Renal Osteodystrophy | Chronic kidney disease | Mixed | Depends on type | ↓ or normal | Mixed lesions, osteosclerosis |
| Fluorosis | Excess fluoride | ↑ or normal | Dense but abnormal | Normal | Joint rigidity |
Paget’s Disease of Bone (Osteitis Deformans)
Overview:
-
Chronic bone disease characterized by both osteolysis and osteosclerosis (excessive bone formation)
-
More common in males over 50 years.
-
Can be monostotic (single bone) or polyostotic (multiple bones).
Etiology:
-
Possible slow-virus infection (paramyxovirus) of osteoclasts.
-
Genetic predisposition:
-
Autosomal dominant inheritance
-
Gene on chromosome 18q → RANK (Receptor Activator of Nuclear Factor κB)
-
Clinical Features:
-
Monostotic: Often asymptomatic.
-
Polyostotic: Pain, fractures, skeletal deformities, possible sarcoma (arise from mesenchymal (connective) tissues).
-
Biochemical marker: Elevated alkaline phosphatase, normal/high calcium.
Morphologic Stages:
-
A. Osteolytic Phase:
-
Excessive bone resorption by large, multinucleated, abnormal osteoclasts.
-
Bone becomes weakened and porous.
B. Mixed Osteolytic-Osteoblastic Phase:
-
Active osteoblasts attempt to lay down new bone.
-
However, this bone is poorly organized.
-
This phase is often the longest.
C. Quiescent (Sclerotic) Phase:
abnormally dense and hardened
-
Osteoclastic and osteoblastic activity subsides.
-
Bone becomes dense but disorganized, mechanically weak, and prone to fractures.
-
4. Tumour-like Lesions of Bone
These are non-neoplastic but mimic bone tumors. Common types:
A. Fibrous Dysplasia
-
Developmental anomaly.
-
Replacement of normal bone with fibrous tissue + irregular trabeculae of woven bone (Immature, disorganized bone. Randomly oriented collagen fibers, Haphazard arrangement of trabeculae, High cellularity (many osteocytes)
Types:
-
Monostotic (70% cases): Solitary bone, usually ribs, craniofacial bones, femur, tibia.
-
Polyostotic (25%): Multiple bones
-
Albright Syndrome (McCune-Albright):
-
Polyostotic fibrous dysplasia
-
Café-au-lait spots (caused by increased melanin)
-
Sexual precocity & endocrine abnormalities (more common in females)
-
Morphology:
-
Gross: 2–5 cm lesions in cancellous bone, smooth cortex.
-
Microscopy: Fibroblastic stroma with irregular, curved trabeculae (fish-hook/Chinese letter pattern). No osteoblastic rimming.
B. Fibrous Cortical Defect (Metaphyseal Fibrous Defect)
-
Common in children.
-
Affects metaphyseal cortex of tibia or femur.
Radiology: Eccentric lesion with sharp margins.
Morphology:
-
Gross: Small (<4 cm), granular, brown.
-
Microscopy: Storiform fibrous tissue, osteoclast-like giant cells, haemosiderin-laden macrophages, foamy histiocytes (macrophages with a vacuolated cytoplasm due to the accumulation of lipids, cholesterol, or other metabolic products)

BONE TUMOURS
General Overview
-
Incidence: Comparatively infrequent but clinically significant, particularly due to potential malignancy.
-
Classification:
-
Primary: Arise from the bone itself.
-
Metastatic: Secondary spread from other organs.
-
2. Classification of Bone Tumors
A. Primary Bone Tumors
Tumors that originate directly from bone tissue or its components (osteoblasts, chondrocytes, marrow cells, etc.)
Type Examples Benign/Malignant Osteogenic Osteoid osteoma, Osteoblastoma, Osteosarcoma Both Chondrogenic Enchondroma, Chondroblastoma, Chondrosarcoma Both Fibrous Fibrous dysplasia, Fibrosarcoma Both Vascular Hemangioma, Angiosarcoma Both Hematopoietic Multiple Myeloma, Lymphoma Malignant Other Ewing sarcoma, Giant cell tumor Usually malignant or locally aggressive B. Metastatic Bone Tumors
Tumors that spread to the bone from a primary malignancy elsewhere in the body.
-
Common primary sources:
-
Breast
-
Prostate
-
Lung
-
Kidney
-
Thyroid
-
-
Clinical clue: In adults over 40, bone lesions are more likely metastatic than primary.
3. Diagnosis of Bone Tumors
Accurate diagnosis requires multi-modality correlation:
A. Clinical Examination
-
History: Pain, swelling, pathological fracture
-
Physical exam: Palpable mass, tenderness, deformity, neurovascular compromise
B. Radiological Imaging
Modality Purpose X-ray First-line; identifies location, size, type of lesion (lytic/blastic/mixed) CT scan Detailed bony architecture, cortical involvement MRI Soft tissue extension, marrow involvement, skip lesions Bone scan (Tc-99m) Detects multifocal disease or metastasis PET-CT For staging of malignant tumors and response assessment C. Histopathology (Biopsy)
-
Core needle biopsy or open biopsy is essential for:
-
Confirming tumor type
-
Grading and staging
-
Planning surgery or chemotherapy
-
-
Histological features: Cell type, mitosis, matrix (osteoid, chondroid), necrosis, giant cells
D. Biochemical Markers
Marker Use Serum calcium ↑ in bone resorption or metastasis (e.g., myeloma, breast CA) Serum phosphate Altered in metabolic bone disease Alkaline phosphatase ↑ in bone turnover (e.g., osteosarcoma, Paget’s disease) Acid phosphatase ↑ in prostate carcinoma with bone metastases E. Special Tests for Specific Tumors
Tumor Type Special Test Multiple Myeloma Serum/urine protein electrophoresis (M-protein), Bence-Jones proteins Neuroblastoma Urinary catecholamines (VMA, HVA) Lymphoma/Leukemia Bone marrow aspiration/biopsy Benign Bone-Forming Tumours
-
1. Osteoma
-
Nature: Rare, benign, slow-growing
-
Sites: Flat bones of the skull and face; may extend into paranasal sinuses or orbit.
-
Associations: May be part of Gardner’s syndrome.
-
Radiology: Dense, ivory-like bony mass.
-
Microscopy: Mature lamellar bone trabeculae with fibrovascular stroma.
2. Osteoid Osteoma
-
Age: Young individuals, <30 years.
-
Size: Small (<1 cm).
-
Location: Cortex of long bones.
-
Symptoms: Painful (often nocturnal, relieved by NSAIDs).
-
Radiology: Central radiolucent nidus with dense sclerotic surrounding bone.
3. Osteoblastoma
-
Age: Adolescents and young adults.
-
Size: Larger (>1 cm).
-
Location: Medullary cavity—vertebrae, ribs, ilium, long bones.
-
Symptoms: Painless.
-
Radiology: No sclerotic rim.
-
Histology: Similar to osteoid osteoma; difficult to distinguish microscopically.
Malignant Bone-Forming Tumour
Osteosarcoma (Osteogenic Sarcoma)
-
Most common primary malignant bone tumour.
-
Age: 10–20 years (primary); >40 years in secondary cases.
-
Sex: M > F.
-
Site: Metaphysis of long bones:
-
Around the knee (distal femur, proximal tibia) – 60%
-
Proximal humerus – 10%
-
Pelvis & proximal femur – 15%
-
Rare: jaws, vertebrae, skull, soft tissues (extraskeletal osteosarcoma)
-
Classification by Pathogenesis
-
Primary Osteosarcoma:
-
No predisposing condition.
-
Etiology: Likely genetic and environmental.
-
RB gene mutation (retinoblastoma link)
-
p53 mutation
-
MDM2, cyclin D1, p16, CDK4 alterations
-
Exposure to radiation, viruses.
-
-
-
Secondary Osteosarcoma:
-
Develops on pre-existing lesions:
-
Paget’s disease, fibrous dysplasia, chronic osteomyelitis, infarcts, fractures.
-
-
More aggressive.
-
Pathology
-
Location: Metaphyseal region, starts in medulla.
-
Spread: Expands to cortex, periosteum, and surrounding soft tissues.
-
Epiphyseal plate may delay spread.
-
Radiology:
-
Sunburst appearance (spiculated bone formation)
-
Codman’s triangle (periosteal lifting)

-
Clinical:
-
Pain, swelling, tenderness.
-
Raised serum alkaline phosphatase; normal Ca²⁺ and PO₄³⁻.
-
-
Metastasis:
-
Via bloodstream.
-
Commonly to lungs, other bones, brain.
-
Cartilage-forming Tumors and Giant Cell Tumors of Bone
1. Enchondroma
-
Definition: A benign cartilage-forming tumor located within the medullary cavity of bone.
-
Common Sites: Long bones and ribs.
-
Age/Sex: Any age, both sexes.
-
Clinical Features:
-
Often asymptomatic.
-
May present with pain or pathological fractures.
-
-
Radiology: Radiolucent, lobulated lesion with spotty calcifications.
-
Malignant Potential:
-
Solitary lesions rarely become malignant.
-
Multiple enchondromas (enchondromatosis) may transform into chondrosarcoma.
-
-
Morphology:
-
Gross: Translucent lobulated mass in medullary cavity.
-
Microscopy: Lobules of mature hyaline cartilage separated by vascular fibrous stroma ± calcification.
-
Differentiation: No tissue invasion or cellular atypia (distinguishes it from chondrosarcoma).
-
2. Chondroblastoma
-
Definition: A rare benign cartilage-producing tumor of the epiphysis.
-
Common Sites: Around knee (upper tibia, lower femur), upper humerus.
-
Age/Sex: <20 years, male > female (2:1).
-
Radiology: Sharply circumscribed lytic lesion (area of bone destruction seen on imaging , where normal bone has been resorbed or replaced, leading to a radiolucent (dark) appearance.) with foci of calcification.
-
Clinical Features: Local pain, tenderness, discomfort; may recur after curettage (curettage is frequently used for the removal of benign bone tumors or cysts).

-
Morphology:
-
Gross: Epiphyseal mass up to 5 cm, soft, chondroid (tissue that resembles cartilage—specifically hyaline cartilage) with hemorrhage, necrosis, calcification.
-
Microscopy:
-
Highly cellular with small, round to polygonal chondroblasts.
-
Multinucleated osteoclast-like giant cells.
-
Chondroid matrix and focal calcification.
-
-
3. Chondromyxoid Fibroma
-
Definition: An uncommon benign tumor of immature cartilage.
-
Common Sites: Metaphysis of long bones (e.g., upper tibia, lower femur).
-
Age/Sex: 2nd–3rd decade, male > female.
-
Radiology: Well-demarcated radiolucent lesion with some calcification and bone expansion.
-
Clinical Features: Pain, swelling, discomfort; may recur after curettage.
-
Morphology:
-
Gross: Gray-white, lobulated mass <5 cm, in metaphysis, often with sclerotic rim (increased bone density or hardening, typically seen on radiologic imaging as a white or bright (radiopaque) area).
-
Microscopy:
-
Lobules separated by fibrous tissue and giant cells.
-
-
4. Chondrosarcoma
-
Definition: A malignant cartilage-producing tumor.
-
Types:
-
Central (primary): Arises within the medulla.
-
Peripheral: Arises in cortex/periosteum; can be secondary to benign tumors like osteochondroma or enchondroma.
-
-
Sites: Central skeleton – pelvis, ribs, shoulders; also around knee.
-
Age/Sex: 30–60 years, male > female.
-
Radiology: Large, osteolytic mass with calcification.
-
Clinical Features: Slow-growing mass with pain and swelling; metastasizes in higher grades (lungs, liver, kidney, brain).
-
Morphology:
-
Gross: Large, lobulated, firm mass; bluish-white, gelatinous with ossification.
-
Microscopy:
-
Invasive growth of lobules of anaplastic cartilage cells (Anaplastic refers to cells that have lost their normal structure and function).
-
Cytologic malignancy: hyperchromatism, pleomorphism, multinucleation, tumor giant cells (reflects abnormal cell fusion or proliferation)
-
-
5. Giant Cell Tumor (Osteoclastoma)
-
Definition: Aggressive, potentially malignant tumor with numerous osteoclast-like giant cells.
-
Sites: Epiphysis of long bones – distal femur, proximal tibia, distal radius, upper fibula.
-
Age/Sex: 20–40 years, both sexes.
-
Radiology: Osteolytic lesion with "soap bubble" appearance.
-
Clinical Features: Pain (on movement/weight-bearing), swelling, pathological fracture.
-
Morphology:
-
Gross: Eccentric epiphyseal lesion, hemorrhagic and necrotic with cystic changes.
-
Microscopy:
-
Numerous multinucleate giant cells (~100 nuclei).
-
Stromal mononuclear cells are the true tumor cells – round/spindle, mitotically active.
-
Stroma is vascular with scant collagen, hemorrhage, and macrophages.
-
-
-
Biologic Behavior:
-
Recurrent in 40–60% cases after curettage.
-
4% metastasize to lungs (histologically benign).
-
May transform into sarcoma (especially post-radiotherapy).
-
-
Differential Diagnosis: Brown tumor, aneurysmal bone cyst.
-
Cell of Origin: Likely mesenchymal stromal cells; giant cells formed by monocyte fusion facilitated by TGF-β.
6. Ewing’s Sarcoma
-
Definition: Highly malignant small round cell tumor of neuroectodermal origin.
-
Variants:
-
Skeletal (classic) Ewing’s sarcoma.
-
Soft tissue Ewing’s sarcoma.
-
Primitive neuroectodermal tumor (PNET).
-
-
Age/Sex: 5–20 years, female > male.
-
Genetics: Translocation t(11;22)(q24;q12) – common to all 3 variants.
-
Sites: Arises in medullary cavity of diaphysis or metaphysis of long bones.
JOINTS
Types of Joints
-
Diarthrodial (Synovial) Joints:
-
Have a joint cavity.
-
Ends of two bones are held by a joint capsule reinforced by ligaments and tendons.
-
Articular surfaces covered by hyaline cartilage, thicker in weight-bearing areas.
-
Joint cavity lined by synovial membrane (synovium) which produces synovial fluid for lubrication.
-
-
Synarthrodial (Nonsynovial) Joints:
-
No joint cavity.
-
Less commonly affected by diseases.
-
Synovial Membrane Structure
-
Two layers:
-
Inner layer: 1–4 cells thick made of synoviocytes (2 types on EM):
-
Type A synoviocytes: Macrophage-like, produce degradative enzymes.
-
Type B synoviocytes: Fibroblast-like, synthesize hyaluronic acid.
-
-
Outer layer: Loose, vascular connective tissue.
-
OSTEOARTHRITIS (OA) / DEGENERATIVE JOINT DISEASE (DJD)
Definition
-
The most common chronic disorder of synovial joints, characterized by progressive degeneration of articular cartilage especially in weight-bearing joints.
Types and Pathogenesis
-
Primary OA
-
Occurs in elderly (starting after 40 years), more common in women.
-
Etiology unclear; associated with wear and tear, minor trauma, aging, obesity, heredity.
-
Genetic susceptibility (e.g., mutations in FRZB gene affecting cartilage growth).
-
-
Secondary OA
-
Can occur at any age due to previous joint damage: injury, fracture, inflammation.
-
-
Molecular Mechanism
-
Cartilage breakdown mainly due to degradation of type II collagen.
-
Mediated by cytokines like IL-1, TNF, and nitric oxide.
-
Morphologic Features of OA
-
Articular Cartilage
-
Cartilage loosens, flakes, fissures → fragments break off exposing subchondral bone.
-
Radiology: Narrowed joint space due to cartilage loss.
-
-
Bone Changes
-
Death of superficial osteocytes, increased osteoclast activity.
-
Bone remodeling causes flattening and mushroom-like shape of joint ends.
-
Loose osteophytes may form joint mice (loose bodies).
-
-
Synovium
-
Initially normal.
-
Advanced cases show low-grade chronic synovitis.
-
Possible synovial effusion.
-
Commonly Involved Joints
-
Weight-bearing joints: hips, knees, vertebrae.
-
Interphalangeal joints of fingers also affected.
Clinical Features
-
Joint stiffness, reduced mobility, pain—worse after rest (morning stiffness).
-
Enlarged bony nodules on distal finger joints called Heberden’s nodes (common in women, hereditary tendency).
-
In spine, osteophytes may compress nerve roots → pain, spasms (sudden, involuntary contraction of a muscle or group of muscles), neurological signs.

Rheumatoid Arthritis (RA)
Overview
-
Chronic autoimmune disease of unknown exact cause.
-
Predominantly causes inflammatory arthritis, usually symmetrical and mainly affects peripheral joints.
-
Systemic effects may involve hematologic, pulmonary, neurological, and cardiovascular systems.
| System | Common Manifestations |
|---|---|
| Hematologic | Anemia, thrombocytosis, Felty’s syndrome |
| Pulmonary | Pleuritis, nodules, pulmonary HTN |
| Neurological | neuropathies |
| Cardiovascular | Atherosclerosis, pericarditis, myocarditis |
-
Peak incidence: 3rd to 4th decades of life. (Genetic Susceptibility, Immune Dysregulation, Hormonal Factors, Environmental Triggers).
-
Female predominance: 3–5 times more common in females.
-
Strong genetic association with HLA-DR4 and HLA-DR1.
-
Familial aggregation common.
Clinical Features
-
Insidious onset: fatigue, weakness, joint stiffness, arthralgia, and myalgia.
-
Followed by symmetrical joint pain and swelling.
-
Most commonly involved joints: hands, wrists, feet.
-
Unlike rheumatic fever’s migratory arthritis, RA arthritis persists in involved joints.
-
About 20% develop rheumatoid nodules (extensor surfaces of elbows, fingers).
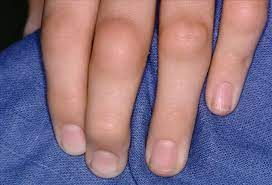
-
Approximately 80% are seropositive for rheumatoid factor (RF) (an autoantibody—usually of the IgM class—that targets the Fc portion of IgG antibodies).
-
RF can be elevated in other diseases: viral hepatitis, cirrhosis, leprosy.
-
Advanced radiological features: joint space narrowing, ulnar deviation of fingers, radial deviation of wrists.
-
Labs: mild normocytic normochromic anemia, elevated ESR, mild leukocytosis, hypergammaglobulinemia.
Etiopathogenesis
Immunologic Factors
-
Autoimmune phenomenon with production of rheumatoid factor (RF) (IgM and IgG).
-
Circulating immune complexes (IgG-RF).
-
Other autoantibodies: ANA, anti-collagen type II, anti-cytoskeleton (against proteins of the cell’s cytoskeleton, which includes actin filaments, intermediate filaments, and microtubules)
-
Inflammatory infiltration predominantly by CD4+ T cells, macrophages in synovium.
Trigger Factors
-
Infectious agents suspected: mycoplasma, EBV, CMV, rubella virus.
-
Genetic predisposition (HLA-DR4, HLA-DR1) important.
-
Superantigens from microorganisms may activate immune response.
Immunopathogenesis (Simplified Model)
-
Antigenic trigger in genetically predisposed (HLA-DR) individual → activation of CD4+ T cells.
-
T cells release cytokines: TNF-α, IFN-γ, IL-1, IL-6.
-
Cytokines activate endothelial cells, B cells, macrophages.
-
B cells produce RF (IgM anti-IgG antibody).
-
Immune complexes cause inflammation → synovium, vessels, collagen damaged.
-
Macrophages release more cytokines → pannus formation (vascularized granulation tissue).
-
Pannus destroys cartilage and bone → fibrosis → joint deformities.
Morphologic Features
Articular Lesions
-
Initially affects small joints of hands and feet; symmetric.
-
Involves wrists, elbows, ankles, knees; cervical spine commonly affected.
-
Histology: proliferative synovitis with pannus formation.
-
Features:
-
Villous hypertrophy of synovium.
-
Thickened synovial membrane (edema, congestion).
-
Dense mononuclear infiltrate (lymphocytes, plasma cells, macrophages).
-
Fibrinoid necrosis and fibrin deposits.
-
-
Pannus invades cartilage and bone → cystic resorption, demineralization.
-
Tendons may weaken or rupture.
Extra-Articular Lesions
-
Vasculitis affecting vessels, lungs, pleura, pericardium, myocardium, nerves, eyes.
-
Rheumatoid nodules: subcutaneous, especially over pressure points (elbows, occiput, sacrum).
-
Central fibrinoid necrosis surrounded by epithelioid macrophages and lymphocytes.
-
Similar nodules may occur in lungs, pleura, heart valves, other organs.
-
Variants of RA
-
Juvenile RA:
-
Onset under 16 years.
-
Acute fever, mainly knees and ankles.
-
RF rarely present.
-
-
Felty’s Syndrome:
-
RA with splenomegaly and hypersplenism.
-
Leads to hematologic abnormalities.
-
-
Ankylosing Spondylitis (Rheumatoid spondylitis):
-
Involves sacroiliac joints, spine.
-
More common in young males.
-
Strong association with HLA-B27.
-
May be linked with inflammatory bowel disease, anterior uveitis, Reiter’s syndrome (Triad of arthritis, urethritis, conjunctivitis).
-
SKELETAL MUSCLES
Normal Structure

-
Striated skeletal muscles are made up of bundles called fascicles.
-
Each fascicle is surrounded by perimysium, a connective tissue sheath containing blood vessels and nerves.
-
Each individual muscle fiber within a fascicle is enveloped by a delicate connective tissue called endomysium.
-
A muscle fiber is a long, multinucleated cell (~100 μm diameter, several cm length).
-
Nuclei are located peripherally beneath the sarcolemma (muscle fiber plasma membrane).
-
Cytoplasm contains myofilaments:
-
Thick filaments: Myosin
-
Thin filaments: Actin
-
-
Arrangement of myofilaments produces cross-striations seen under light microscopy.
-
Functional unit: Sarcomere (distance between two Z bands), composed of:
-
Central A band (anisotropic)
-
Lateral I band (isotropic)

-
Functions:
-
Convert chemical energy → mechanical energy (contraction)
-
Store energy and proteins (Glycogen (stored glucose) and Phosphocreatine (rapid ATP source)
-
Play a role in body metabolism (glucose metabolism, thermogenesis)
-
-
Muscle contraction requires nerve supply, with each motor unit including:
-
Motor neuron cell body (spinal cord anterior horn or cranial nerve nucleus)
-
Axon of motor neuron
-
Neuromuscular junction
-
Innervated muscle fibers (muscle cells (fibers) that receive signals from a motor neuron through a structure called the neuromuscular junction (NMJ))
-
-
Contraction initiated by acetylcholine release across synaptic gap, generating muscle fiber action potential.
Diseases Affecting Skeletal Muscles
Neurogenic Diseases
-
Characterized by muscle weakness and fatiguability.
-
Common examples:
-
Myasthenia gravis (MG)
-
Congenital myasthenia
-
Eaton-Lambert syndrome (associated with lung carcinoma)
-
Denervation atrophy
-
Myasthenia Gravis (MG)
-
Autoimmune disorder damaging acetylcholine receptors (AChR) at motor end-plates.
-
More common in adult women (3:2 ratio).
-
Symptoms: weakness starting with ocular muscles → spreading to trunk & limbs.
-
Mortality ~10%, mainly due to respiratory muscle involvement.
-
Associated with other autoimmune diseases: autoimmune thyroiditis, rheumatoid arthritis, SLE, pernicious anemia.
-
Pathogenesis:
-
Reduced AChRs and flattened postsynaptic folds → impaired neuromuscular transmission.
-
Autoantibodies against AChRs block receptor sites.
-
Thymus involvement: thymoma or thymic hyperplasia common; thymectomy may help. (Faulty T-cell selection in the thymus may fail to eliminate AChR-reactive T-cells.)
-
-
Morphology:
-
Grossly normal muscles early; atrophy later.
-
Light microscopy: lymphocytic infiltrates, degenerating fibers.
-
Electron microscopy: reduced synaptic area, fewer AChRs.
-
Immunocytochemistry: IgG and complement deposits at neuromuscular junction.
-
Denervation Atrophy
-
Loss of nerve supply leads to muscle atrophy. Without nerve signals, muscles shrink, weaken, and lose function over time.
-
Features:
-
Small angulated muscle fibers alternating with normal/hypertrophic fibers.
-
May progress to dystrophic changes.
| Dystrophic Feature | Description |
|---|---|
| Muscle fiber necrosis | Dying muscle fibers due to prolonged disuse |
| Fibrosis | Replacement of muscle tissue with connective tissue |
| Fatty infiltration | Muscle replaced by fat cells |
| Regeneration attempts | Muscle tries to regenerate, causing fiber size variation |
| Internal nuclei | Nuclei shift from periphery to center (a dystrophic feature) |
-
Seen in diseases like:
-
Amyotrophic lateral sclerosis (ALS)
-
Peripheral neuropathies
-
Myopathic Diseases (Myopathies)
-
Primary muscle diseases with chronic muscle weakness.
-
Five groups: hereditary (muscular dystrophies), inflammatory, endocrine, metabolic, toxic.
-
Focus on Muscular Dystrophies.
Muscular Dystrophies
-
Genetically inherited, progressive muscular weakness.
-
Six major types with differences in inheritance, onset, symptoms, and course:
| Type | Inheritance | Age of Onset | Clinical Features | Other Systems Involved | Course |
|---|---|---|---|---|---|
| Duchenne’s | X-linked recessive | By age 5 | Symmetric weakness (pelvifemoral), pseudohypertrophy of calves | Cardiomegaly; reduced intelligence | Progressive; death ~20 y |
| Becker’s | X-linked recessive | 2nd decade | Slow progressive weakness (girdle) | Cardiomegaly | Benign variant of Duchenne |
| Myotonic type | Autosomal dominant | Any decade | Weakness + myotonia (eyelids, face, limbs) | Cardiac conduction defects, cataracts | Benign |
| Facioscapulohumeral | Autosomal dominant | 2nd–4th decade | Weakness of face, scapula, humerus | Hypertension | Benign |
| Limb-girdle | Autosomal recessive | Early childhood–adult | Weakness of shoulder and hip girdle | Cardiomyopathy | Variable |
| Oculopharyngeal type | Autosomal dominant | 5th–6th decade | Weakness of extraocular, eyelids, face, pharynx | — | Rarely progressive |
-
Morphology common to all:
-
Muscle fiber necrosis, regeneration.
-
Replacement by fibrosis and adipose tissue.
-
Table 28.4: Classification of Neuromuscular Diseases by Motor Unit Site
| Site of Motor Unit | Disease Examples |
|---|---|
| I. Anterior Horn Cell | Spinal muscular atrophy; Amyotrophic lateral sclerosis (ALS) |
| II. Peripheral Nerve | Carpal tunnel syndrome; Mononeuritis multiplex; Diabetic neuropathy |
| III. Neuromuscular Junction | Myasthenia gravis |
| IV. Muscle | Duchenne’s muscular dystrophy |



Comments
Post a Comment